Journal of Neurology, Neurosurgery, Psychiatry誌 (JNNP) の新着論文をチェックしていたら、偶然 2015年4月26日のブログで紹介した抗 NT5C1A抗体に関する論文が報告されているのを見つけました (2015年4月9日 online published)。
・封入体筋炎患者 25人中 18名 (72%) で抗 NT5C1A抗体陽性だった。
・女性の方が抗体陽性率が高かった (OR=2.30)。
・抗体陽性の方が立ち上がるのに時間を要し、歩行器や車椅子を必要としやすかった。
・抗体陽性の方が嚥下障害が多く、努力性肺活量が減少していた。
・顔面筋力低下は抗体陽性の方が頻度が高かった (50% v.s. 14%)
余談ですが、JNNPも勤務先で契約していないため JAMA neurologyに引き続いて個人契約が必要になったんですよね・・・。引越し費用、家具代、電化製品代 (洗濯機、エアコン)、ピアノ搬送代等々で、4月分のカード請求額が 967,889円だった私にとって、痛い出費です。クイーンサイズのベッドが余計な出費だったか・・・。
2015年2月5日のブログで、多発筋炎と皮膚筋炎の総説を紹介しました。
その総説の supplementary dataには筋炎で陽性となる自己抗体の一覧表があり、孤発性封入体筋炎の約 70%で抗NT5C1A抗体が陽性になると書いてありました。この抗体、どこで測定してもらえるのだろうと思って調べると、熊本大学が検査を受け付けていました。
封入体を欠く封入体筋炎などで、筋生検をしても確定診断を得られないことがあるので、診断の一助に使えそうです。お世話になる機会がありそうです。
Wikipediaで遊んでいたら、、面白いサイトを見つけました。
下ネタっぽい地名とか、人名地名とか、面白いですね。
あと、下記リンクから各地方の難読地名へのリンクがありますが、ほとんど読めませんでした。
JAMA neurologyに興味深い論文が載っていました (2015年4月20日 online published)。
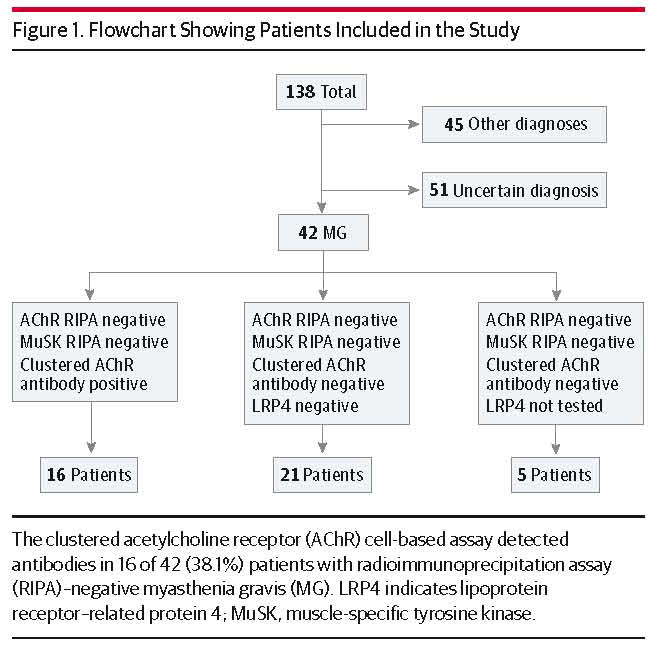
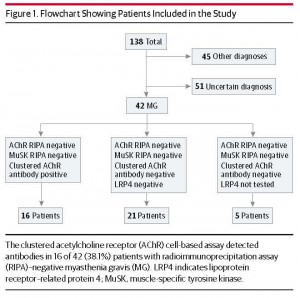
抗AChR抗体陰性 (と抗MuSK抗体) が Radioimmunoprecipitation assay (RIPA) で陰性だった重症筋無力症患者 42名に対して、HEK細胞の表面に AChRを発現させる Cell Based Assayで検査したところ、16名 (38.1%) で抗 AChR抗体が検出できたという報告です。検査法によってここまで抗体の検出率が違うのかと驚きました。この研究の結果を見ると、これまではかなりの患者で抗 AChR抗体が見逃されてきたことになります。もちろん、重症筋無力症の診断は抗体のみでなされる訳ではありませんが。
日本では通常 SRLなどの検査機関で抗 AChR抗体を調べると思います。SRLでの検査法がどうなっているのか調べた所 RIA法でした。この論文のように Cell based assayでも検査できるようになれば、検査の精度が改善するかもしれません。
MG cell based assay
なお、この研究では抗 LRP4抗体も調べています。日本では、以前お伝えしたとおり抗 LRP4抗体は川棚医療センターで測定頂けます。
(参考)
・神経内科のトレンド2011
・autoimmune autonomic ganglionopathy
新しい勤務先では、以前ほど文献が自由に閲覧出来ません。私はお気に入りの医学雑誌を 10誌くらい頻繁にチェックしていて、それらにアクセスできないのはかなりのストレスになります。例えば、JAMA neurologyは現在の勤務先では取り寄せないと読めません。そこで必要な医学雑誌を個人契約し、定期購読することにしました。
JAMA neurology
手続きがてら JAMAのサイトをチェックしていたら、次の一文を見つけました。
発展途上国の研究機関では無料で JAMAにアクセスできるそうです。素晴らしい配慮だなと思いました。
Amazonで購入した金額の合計額を簡単に知る方法があるらしいです。
試しにやってみたら、2002年に医師になってからこれまでで、5850247円でした。計算すると、一ヶ月当たり 35000~40000円くらいになります。もっと CDとか本とか沢山買っているイメージがあったので、意外でした。
ただ、日本で手に入らないマイナーな医学書をイギリスやアメリカのAmazonで買っているので、実際にはもう少し利用していると思います。
Annals of Neurologyの “accepted article” の欄に、興味深い論文が掲載されていました。
OPA1や Mfn2はミトコンドリア外膜に存在しミトコンドリアの融合に関係する蛋白質で、これが分解されると膜電位の維持できない不良なミトコンドリアが健常なミトコンドリアと融合しなくてすむメリットがあります。ミトコンドリアの膜電位が低下すると、これらの蛋白質は parkinによってユビキチン化を受け、プロテアソームやオートファジーによって分解されます。parkinを活性型にするのが PINK1という蛋白質です。
PINK1→(リン酸化) →parkin→ (ユビキチン化) →OPA1, Mfn2・・・・
この中で、PINK1, parkinはパーキンソン病の原因蛋白質として知られていますが、OPA1変異では常染色体優性視神経萎縮となり、Mfn2変異では視神経萎縮を合併した Charcot-Marie-Tooth 病 (CMT2A) となります。OPA1の GTPase domain 変異は視神経萎縮に加えて重度の感音性難聴、小脳失調、軸索性運動感覚ニューロパチー、慢性進行性外眼筋麻痺、ミトコンドリア筋症を含む DOA-plus 症候群を合併することは過去に報告されていましたが、何故 parkinの下流にある蛋白質の変異でパーキンソン病にならないのだろう・・・とこれまで疑問に思っていました。
ところが、OPA1変異でパーキンソニズムを呈した 2家系が、ボローニャとメッシーナから報告されました。両家系の発症した患者は、20 ~ 30 歳代で若年性高血圧、パニック発作を伴う不安症、30 ~40 歳代で緩徐進行性の眼瞼下垂と眼筋麻痺、ミトコンドリア筋症、末梢神経障害、小脳萎縮、失調、感音性難聴を来しました。そして、高齢になるとパーキンソニズムや認知症を合併した患者が 6 人おり、MRI でびまん性皮質萎縮 、DAT scan での異常を伴っていました。いずれの家系でも、著明な視力低下や視神経萎縮を来したのは 1 名ずつに過ぎませんでした。
このように、通常 OPA1変異でみられるはずの常染色体優性視神経萎縮がほとんどみられず、パーキンソニズムが高率に見られたというのは、非常に興味深いことと思います。患者由来の線維芽細胞の解析では、オートファジー (mitophagyを含む) の亢進が示されていましたが、これが発症にどう影響を与えているのかはこれからの問題だと思います。ひょっとすると、GTPase domain近傍の変異という部位も意味を持っているのかもしれませんね。なお、今回の症例では、parkin, PINK1遺伝子に変異がないことは確認されています。また、その他のミトコンドリア遺伝子に変異がないことも確認されています。
ということで、私にとってこの論文のツボは下記の点でした。
・ミトコンドリア異常症が多彩な表現型を示すこと
・parkin-PINK1 の下流に存在する OPA1 の変異でパーキンソニズムが出現したこと
・ミトコンドリア異常と変性疾患の関連が示唆されること
2015年4月9日の New England Journal of Medicine (NEJM) の MGH case recordsが、Neisseria meningitidisによる髄膜炎でした。補体 C8欠損があると、Neisseria属による感染を起こしやすいんですね。以前、ほぼ同じ症例を経験したので、その時のことを思い出しながら読みました。
細菌性髄膜炎について勉強する方には、少し古いですが下記の論文が御勧めです。700例近い細菌性髄膜炎を解析し、それぞれの症状の出現する割合 (例えば、頭痛 87%, 38℃以上の発熱 77%, 皮疹 26%など) や、予後の良い群と悪い群の比較 (例えば、予後の悪い群では髄液細胞数が少ないなど) の記載があり、勉強になります。
2015年3月19日の New England Journal of Medicine (NEJM) の MGH case recordsが、前頭側頭葉変性症 (FTLD) でした。
認知症の診断を進める際の基本的事項と、前頭側頭型認知症について良く纏まっていました。
・初発症状から、最初に脳のどの部位が侵されたかわかる。症状や、脳のどの部位が侵されたかを調べれば、病因が推測できる。
・認知症の鑑別として、うつ病、統合失調症、Creutzfeldt-Jakob病、貧血、ビタミンB12欠乏、甲状腺機能異常、肝障害、腎障害、梅毒、感染症 (Lyme病など), Huntington病、Wilson病、脂質代謝異常症、他の晩発性先天性代謝異常症、白質変性症などが挙げられる。
・前頭側頭型認知症が左脳から発症するときは、原発性進行性失語症を呈する。右脳から発症するときは、精神症状を呈する (behavior variant; 行動型)。”disinhibition”, “apathy”, “loss of sympathy and empathy”, “repetitive behaviors”, “hyperorality”, “loss of executive function” のうち、3つの症状が存在すれば、前頭側頭型認知症の疑い診断を下すことができる。
・前頭側頭型認知症のほとんどは孤発性だが、 10~15%に常染色体優性遺伝を示すものがある。
・前頭側頭型認知症の 3つのサブタイプは、tau, TDP-43, FUSという 3つタイプの封入体に基づく。
・Tauは、しばしばパーキンソニズムを合併するが、ALSは合併しない。一方で、TDP-43や FUSは前頭側頭型認知症と ALSをより合併しやすい。
・Pick病は Tau病理 (tau-3R) を呈する。典型的には 50~70歳代に発症し緩徐に進行する。稀に家族性である。
・前頭側頭型認知症のいくつかの型が、progranulin遺伝子変異と運動ニューロン疾患を伴うものを含め、TDP-43と関連している。前頭側頭型認知症行動型の TDP-43サブタイプは一般的である。発症は通常 70歳代始めである。
・前頭側頭型認知症と ALSで家族性のものはしばしば C9ORF72の 6塩基反復による。この型は、TDP43 type Bと関連している。カリフォルニア大学で治療された常染色体優性の前頭側頭型認知症の 53%にこの変異がみられた。この変異の患者では、前頭側頭型認知症より ALSが一般的で、通常 MAPT変異や progranulin変異の患者より高齢発症である。MAPT変異は浸透率が高い。
・FUSサブタイプは、前頭側頭型認知症の約 5%程度である。FUS変異は、前頭側頭型認知症よりも ALSを発症しやすい。若くして発症する前頭側頭型認知症はしばしば FUSサブタイプである。発症は 30~40歳代で、30%に精神病を合併する。
この論文の症例が前頭側頭型認知症であることは、神経内科医であればひと目と思いますが、表現型から遺伝子を考察していくプロセスが、(それが的中するかどうかは別として) とても勉強になりました。また、論文中に “sagging brain syndrome” という用語が登場しましたが、初耳だったので押さえて置こうと思います。
筋萎縮性側索硬化症 (ALS) の新規治療薬 Tauroursodeoxycholic acid (タウロウルソデオキシコール酸, TUDCA) のパイロット研究の結果が、European Journal of Neurology誌に掲載されました (2015年2月9日 published online)。ウルソデオキシコール酸 (商品名 ウルソ) は胆汁うっ滞、肝機能障害の治療で良く用いますが、タウロ・ウルソデオキシコール酸の名前は初めて聞きました。肝臓で産生される親水性の胆汁酸で、脂肪肝の治療に用いられる薬剤なのだそうです。細胞保護や抗アポトーシス作用があるそうです。
リルゾールを内服中の 34名の ALS患者に、TCDUAないしは プラセボを上乗せした。3ヶ月後に評価し、一次アウトカムは ALSFRS-Rスコアのスロープの 15%以上改善、二次アウトカムは群間比較 (ALSFRS-Rスコア、ALSFRS-Rスコアの線形回帰スロープ、有害事象) とした。両群間の有害事象に差はなく、効果があったのは、TUDCA 87%, プラセボ 43% (P=0.021) だった。ベースラインを調整した試験終了時の ALSFRS-Rは、TUDCA群で有意に高かった (p=0.007)。線形回帰スロープでは、TUDCA群の方が有意に進行が遅かった (P<0.01)。
この結果だけ見るとかなり期待が持てそうですが、まだパイロット研究の段階に過ぎないので、この知見を基に今後の臨床研究につなげていく必要があります。ちなみに、このパイロット研究は、Nature Reviewsでも紹介されているようです。
ALSの新規治療薬の話題でもう一つ。チロシンキナーゼ阻害薬 Masitinib (マシチニブ) の第三相試験が現在行なわれており、FDAから希少疾患薬の指定 (Orphan Drug Designation) を受けたそうです。
AB Science: Masitinib Receives Orphan Drug Designation for Amyotrophic Lateral Sclerosis from FDA
良い結果が出て欲しいですね。Masitinibは、ネットで検索すると犬や猫の肥満細胞腫でも用いられている薬剤のようで、薬というのは一見関係なさそうに見える疾患で効果が期待されたりするのが面白いところです。関係なさそうな疾患で効果があった事例としては、パーキンソン病に対する抗てんかん薬ゾニサミド、びまん性汎細気管支炎に対するマクロライド系抗菌薬など、枚挙に暇がありません。